


Seventy years ago, sickle-cell disease was at the cutting edge of biomedical research as the first medical condition to be linked to a molecular cause. But the ensuing decades saw little progress in terms of clinical care, leaving patients afflicted with severe pain and dramatically shortened life expectancy.
“There was a long period of time without any types of treatment,” says Vence Bonham, leader of the Health Disparities Unit at the US National Human Genome Research Institute in Bethesda, Maryland. Indeed, the first sickle-cell drug was approved only in 1998.
Recent progress in gene therapy is now giving researchers the tools they need to tackle this disease at its molecular roots. Several clinical trials have demonstrated the therapeutic promise of manipulating the genome using viruses to deliver genes or CRISPR-Cas9 gene-editing technology to counteract the damage wrought by the mutation that leads to sickle-cell disease.
And, crucially, these technologies can be incorporated into existing protocols for a potent treatment called haematopoietic stem cell (HSC) transplantation – the curative impact of which is limited by a shortage of eligible donors.
“A perfect storm of things on the scientific and technology front has made sickle-cell disease a good proof of principle,” says Mitchell Weiss, a haematologist at St Jude Children’s Research Hospital in Memphis, Tennessee. At least 10 trials of gene therapy are now under way, and early data from dozens of patients indicate the potential for long-term disease control and greatly improved quality of life.
These are still early days, and there are lingering questions about the safety of this approach. Notably, two cancer diagnoses in participants of one trial have left the field on edge as researchers work to determine the roots of these malignancies.
But despite these concerns, the efficacy data have left many experts bullish about the long-term potential of gene therapy.
This includes the tantalising possibility of directly repairing the disease at its genomic source: that is, a true cure. “We don’t use the ‘c word’, but they’re looking really promising,” says Donald Kohn, a stem-cell biologist at the University of California, Los Angeles.
Historically, sickle-cell disease claimed many lives in childhood. Advances in medical care mean that people who are affected can now survive to middle age, and a growing arsenal of drugs is helping these individuals to manage their pain and other symptoms effectively.
But right now, the only option for long-term survival is transplantation with HSCs – the bone-marrow-based progenitors of all blood cell types – from a healthy and immunologically compatible donor.
HSC transplantation can be curative, but there’s one main obstacle: the difficulty in finding well-matched donors. Most people with sickle-cell disease have African, Middle Eastern or South Asian ancestry – ethnicities that are heavily under-represented in donor registries.
“The likelihood to find a matched, unrelated donor is below 20 per cent,” says Selim Corbacioglu, a haematologist at University Hospital Regensburg in Germany. This leaves many patients without options.
Gene therapy solves this problem by turning each patient into their own perfectly matched HSC donor. After treatment with a drug that stimulates the release of HSCs from the marrow into the circulation, the cells are harvested and genetically modified in the laboratory. The person then receives chemotherapy drugs that kill off their remaining natural HSCs.
This creates room for the re-implantation of modified stem cells, while culling HSCs that might continue to produce sickle-shaped red blood cells. Importantly, this need not be a complete replacement — transplantation studies have shown that an infusion comprising just 20% healthy HSCs is sufficient to reverse the disease.
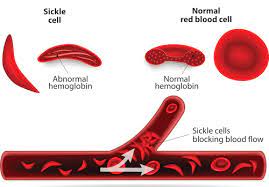
Sickle-cell disease presents a near-ideal opportunity to tap the power of gene therapy because the disorder typically arises from a mutation in a single nucleotide in one gene. That gene encodes the protein β-globin – a key component of haemoglobin, the molecule that red blood cells use to bind and transport oxygen.
The mutated protein misfolds and assembles into fibrous aggregates, resulting in deformed red blood cells that cause painful and damaging obstructions in blood vessels.
Current gene-therapy strategies use two distinct tactics to overcome the effects of this mutation. One restores expression of the fully functional β-globin gene. This can be done by delivering a new copy of the gene to another site in the HSC genome, or by correcting the mutated gene itself.
The other approach is to instead restore expression of a different, naturally occurring protein called γ-globin. This has the same function as β-globin, but is normally produced only during foetal development, where it helps to ensure steady blood oxygenation for the baby in the womb.
In the months after birth, the gene that makes this protein – also called foetal haemoglobin – is permanently switched off in most people.
In 2008, stem-cell biologist Stuart Orkin’s team at Harvard Medical School in Boston, Massachusetts, determined that a protein called BCL11A directly inhibits postnatal production of foetal haemoglobin, and researchers have devised genetic interventions that target BCL11A to lift this inhibition. Fortuitously, reactivation of the foetal protein also downregulates production of adult β-globin.
“You’re just reversing the switch,” says David Williams, chief of haematology and oncology at Boston Children’s Hospital in Massachusetts. In the context of sickle-cell disease, production of the foetal protein is even beneficial: “It actually has anti-sickling characteristics that are very effective,” says Williams.
Strategies based on restoring functional β-globin expression have generally relied on lentiviruses to deliver their therapeutic payload. These retroviruses can insert foreign genes into host genomes in a process known as transduction.
This cargo could be a normal human β-globin gene, but most researchers are stacking the deck by using engineered gene variants that also prevent aggregation of the sickle form of the protein. This is helpful, given that the mutated gene continues to be expressed alongside the therapeutic gene.
- A Nature report

About author
